Квалификация помещений по GMP

Для чего нужна аттестация чистых помещений на фармпроизводстве?
В соответствии с "Правилами организации производства и контроля качества лекарственных средств", утвержденными Приказом Минпромторга России от 14.06.2013 N 916, и ГОСТ Р 52249-2009, производство лекарственных средств и фармацевтических субстанций должно вестись в чистых помещениях.
При производстве лекарственных средств особое значение имеет понятие "стерильность", означающие "отсутствие живых микроорганизмов". Для обеспечения стерильности на фармпроизводстве технологические операции при производстве лекарственных препаратов, как проходящих финишную стерилизацию, так и производимых в асептических условиях, должны производиться в чистых помещениях или чистых зонах.
Основные требования к чистым помещениям на фармацевтическом производстве
Требования к чистым помещениям для асептического фармпроизводства и производства лекарственных средств, которые могут быть подвергнуты финишной стерилизации, отличаются. Технологический процесс розлива / наполнения (критический процесс) при производстве лекарственных средств, не подвергаемых финишной стерилизации в упаковке, требует чистой зоны "А", окруженной чистой зоной класса "В", чтобы свести к минимум риск контаминации готовой продукции частицами и микроорганизмами.
Для фармацевтической продукции, которая проходит финишную стерилизацию, "Правилами организации производства и контроля качества лекарственных средств" установлены менее жесткие требования, в частности, наполнение продуктами, подлежащими финишной стерилизации, может проводиться в производственной среде класса С, однако, при повышенном риске контаминации (если операции наполнения проходят медленно или упаковки имеют широкое горло, или их необходимо держать открытыми более нескольких секунд до герметизации), наполнение так же должно проводиться в чистой зоне класса А (но с окружающей средой, по крайней мере, класса С).
Требования к оснащенному и эксплуатируемому состоянию должны быть установлены для каждого чистого помещения или комплекса чистых помещений.
| Асептическое производство | |
|---|---|
| Тип зоны | Выполняемые операции |
| А |
Асептическое приготовление и наполнение |
| В |
Зоны, окружающие зону «А» |
| С |
Приготовление растворов для фильтрации |
| D |
Операции с материалами после мойки |
| Операции с продукцией, подлежащей финишной стерилизации | |
|---|---|
| Тип зоны | Выполняемые операции |
| А |
Операции с продуктом, когда его нельзя подвергать риску загрязнения |
| В |
Зоны, окружающие зону «А» |
| C |
Наполнение продуктом |
| D |
Приготовление растворов и подготовка первичной упаковки, материалов и др. для последующего наполнения |
Классы чистоты помещений в фармацевтическом производстве
ГОСТ Р 52249-2009 "Правила производства и контроля качества лекарственных средств" определяет типы чистых зон (А, B, С, D) и соответствующие им классы чистоты по ИСО (ГОСТ Р ИСО 14644-1-2017) для различных чистых помещений фармацевтического производства и отдельных технологических процессов.
| Тип чистой зоны | Максимально допустимое число частиц в 1 м3 воздуха при размере частиц, равном или большем | |||
|---|---|---|---|---|
| В оснащенном состоянии | В эксплуатируемом состоянии | |||
| 0,5 мкм | 5,0 мкм | 0,5 мкм | 5,0 мкм | |
| А | 3520 | 20 | 3520 | 20 |
| В | 3520 | 29 | 352000 | 2900 |
| С | 352000 | 2900 | 3520000 | 29000 |
| D | 3520000 | 29000 | – | – |
Как проводится валидация чистых помещений на фарме?
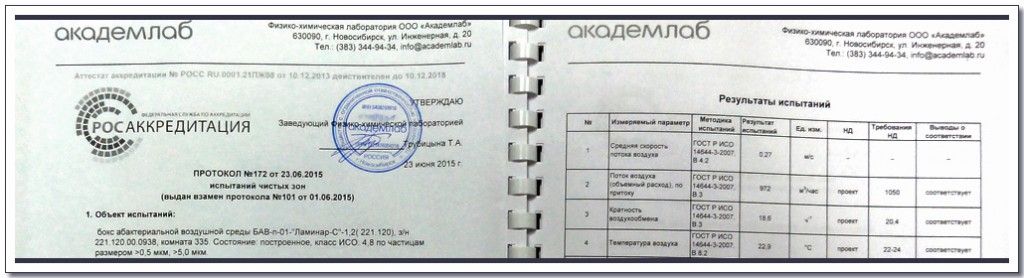
Основным параметром чистого помещения, требующим проверки при проведении аттестации на фармацевтическом производстве, является класс чистоты помещения по ИСО (тип чистой зоны).
Для чистых зон класса А (ИСО 4.8 по частицам с размерами > 5,0 мкм и ИСО 5 по частицам с размерами >0,5 мкм) допустимые концентрации частиц составляют 20 и 3520 шт/м3 соответственно. Для измерения низких концентраций частиц с размерами >5 мкм отбирается не менее 1 м3 воздуха. В ряде случаев может использоваться метод последовательного счета, позволяющий ускорить процесс анализа без ущерба для точности результатов. В частности, этот метод применяется при проверке ламинарных боксов.
В зависимости от особенностей конкретного фармацевтического производства и технологического процесса, проводится также проверка параметров микроклимата (температура и влажность, стабильность поддержания параметров микроклимата), измерение расхода приточного и вытяжного воздуха и кратности воздухообмена и других необходимых параметров.
При наличии в чистой зоне класса "А" однонаправленного потока воздуха, измеряется скорость воздушного потока и оценивается его равномерность. Скорость однонаправленного потока в соответствии с правилами GMP должна лежать в пределах 0,36-0,54 м/с. В закрытых изолирующих устройствах и ламинарных боксах допустим однонаправленный поток воздуха с меньшей скоростью, при этом проверка проводится на соответствие скорости и расхода воздуха технической документации (проектной документации или паспорту бокса).
При аттестации в оснащенном и эксплуатируемом состоянии может также проводиться визуализация воздушных потоков вблизи оборудования, показывающая влияние выступающих частей оборудования на движение воздуха и позволяющая оценить риск возникновения застойных зон.
Еще одним важным показателем, требующим проверки, является целостность финишных HEPA-фильтров. При наличии утечек в самих фильтрах или их уплотнении достижение помещением требуемого класса чистоты может быть затруднено, риск загрязнения продукции резко возрастает.
Испытание фильтров на утечку проводится с использованием генератора аэрозольных частиц и позволяет локализовать утечку и заменить или отремонтировать поврежденные фильтры. При проведении работ по проверке HEPA-фильтров в ходе аттестации чистых помещений на фармпроизводстве специалисты лаборатории Академлаб сразу же информируют технолога и менеджера по качеству фармпроизводства о выявленных утечках, что позволяет заменить или отремонтировать фильтры на месте и сразу провести повторные испытания, что экономит время и средства.
Для чего привлекать к аттестации чистых помещений стороннюю аккредитованную лабораторию?
При наличии у собственной службы качества всего необходимого оборудования для проведения мониторинга и аттестации чистых помещений, привлечение сторонней лаборатории дает дополнительные преимущества и массу новой информации:
- Возможность проведения сравнительных испытаний
- Подтверждение компетентности собственной службы качества
- Наличие протоколов независимой аккредитованной лаборатории, подтверждающих соответствие чистых помещений требования GMP, снимающих множество вопросов в ходе внешних инспекций
Кроме того, фармацевтическая система качества по ГОСТ Р 52537-2006 "Производство лекарственных средств. Система обеспечения качества. Общие требования" регламентирует проведение внутренних аудитов с привлечением независимых лабораторий:
"Аудит может выполняться <...> с привлечением, при необходимости, аккредитованных Испытательных лабораторий для проверки соответствия установленным требованиям (аттестации) оборудования, чистых помещений и процессов".
Отчет об испытаниях чистых помещений на фармацевтическом производстве
Отчет о проведенных испытаниях чистых помещений фармацевтического предприятия включает в себя протоколы измерений и информацию о соответствии помещений требованиям GMP и проектной документации, а также, при необходимости, дополнительную информацию - первичные данные измерений, данные визуализации потоков и т.п.
Оставьте заявку на проведение
квалификации чистых помещений
Наши специалисты свяжутся с Вами в ближайшее время,
проконсультируют по любым исследованиям и ответят на Ваши вопросы
проводим испытания чистых помещений
всех классов чистоты